RAS proteins belong to the small GTPase family. When mutated, they are among the most common oncogenic drivers in cancer. The three RAS isoforms—KRAS, HRAS, and NRAS—have distinct mutation patterns in melanoma: NRAS mutations occur in 15–25% of cases, while KRAS and HRAS mutations are extremely rare. Patients with NRAS-mutant melanoma differ from those with BRAF-activating mutations, typically presenting at an older age and having a history of chronic ultraviolet radiation (UVR) exposure.
Most activating NRAS mutations in melanoma affect codon 61 (e.g., Q61L, Q61K, Q61R). These mutations impair NRAS's GTPase activity, locking it in an active state that activates downstream RAF signaling. The primary pathway driven by oncogenic NRAS is the mitogen-activated protein kinase (MAPK) pathway, which is hyperactivated in most melanomas. This pathway regulates key oncogenic processes: uncontrolled cell growth (via cyclin D1 maintenance and p27KIP1 suppression), enhanced cell survival (through BAD phosphorylation and reduced BIM expression), and cell invasion (by modifying the cytoskeleton).
Oncogenic RAS activation also mediates tumor-induced immune suppression through multiple mechanisms. It upregulates cytokines and chemokines that create an immunosuppressive microenvironment, downregulates tumor-intrinsic interferon signaling, and increases immune checkpoint PD-L1 expression. Specifically, RAS regulates tumor-derived IL-8 (promoting inflammation and progression), CXCR2 ligands (CXCL1, CXCL2, CXCL3, CXCL5), CCL2, and GM-CSF (recruiting and polarizing myeloid-derived suppressor cells [MDSCs] and neutrophils). It also induces IL-10 and TGF-β, which drive regulatory T cell (Treg) differentiation. Impaired interferon signaling reduces major histocompatibility complex (MHC) expression on tumor cells, hindering T cell-mediated recognition. Additionally, RAS reprograms tumor metabolism, leading to lactic acid accumulation that impairs T cell function.
Current standard care for NRAS-mutant melanoma involves immune checkpoint inhibitor (ICI) therapy, such as anti-PD-1, anti-PD-1+LAG-3, or anti-PD-1+anti-CTLA-4. While 30–40% of patients initially respond, 30% of responders later relapse. Patients unresponsive to ICIs or developing resistance have limited therapeutic options. Although RAS was once considered undruggable, inhibitors targeting the inactive (GDP-bound) mutant KRAS (KRAS(OFF)) have shown promising clinical activity. For example, the KRAS G12C inhibitor sotorasib is effective against NRAS G12C-mutant tumors, particularly when combined with EGFR inhibition in colorectal carcinoma.
RMC-6236 is a preclinical tool compound representing oral RAS(ON) multi-selective inhibitors, including the investigational agent RMC-7977. These compounds target active (GTP-bound) NRAS, HRAS, and KRAS (both mutant and wild-type) by binding cyclophilin-A and remodeling its surface to form a high-affinity binary complex with RAS(ON). This complex sterically blocks RAS-effector interactions, inhibiting downstream signaling. This study investigates RMC-6236's antitumor activity in NRAS-mutant melanoma mouse models, focusing on adaptive immunity. Results show durable responses depend on CD4+ and CD8+ T cell activation and reduced MDSC recruitment. Combining RMC-6236 with anti-PD-1 significantly improves survival and achieves complete tumor regression in anti-PD-1-resistant preclinical models. Preliminary clinical responses were observed in two NRAS-mutant melanoma patients treated with daraxonrasib in an ongoing Phase I/Ib trial.
NRAS-Mutant Melanoma Cell Lines (Q61 and G13 Mutations) Are Sensitive to RMC-6236 In Vitro
We first evaluated RMC-6236's activity against 11 NRAS-mutant mouse melanoma cell lines. The compound potently inhibited growth in OSUMMER.1–5, 9–13, SW1 cells, and the human IPC-298 cell line. Three-week colony formation assays confirmed durable growth inhibition in OSUMMER.13, SW1, and IPC-298 cells, with RMC-6236 inducing apoptosis in OSUMMER.13 and SW1 cells.
Phospho-ERK inhibition dynamics showed rapid ERK signaling suppression in OSUMMER.13 cells (with partial recovery by 4 h), slower recovery in SW1 and IPC-298 cells, and cell line-dependent effects on phospho-AKT (increases in OSUMMER.13, decreases in SW1, and biphasic changes in IPC-298). RTK array analysis revealed model-specific adaptations: SW1 cells showed increased EGFR/NGFR phosphorylation; OSUMMER.13 cells had minor ERBB3 changes; IPC-298 cells exhibited upregulated insulin receptor (8 h) and TIE1 (48 h) phosphorylation, with reduced NGFR, c-MET, and EphA1 (48 h).
RMC-6236 also enhanced tumor immune recognition, increasing MHC Class I (H2Db, H2Kb), MHC Class II (I-A/I-E), and PD-L1 expression in a dose-dependent manner. Cytokine secretion was modulated: OSUMMER.13 cells showed increased TNFRSF11B, MMP-2, and WISP-1, and decreased M-CSF, GM-CSF, CCL2, CXCL1, CXCL12, and TIMP-1. SW1 cells exhibited reduced TIMP-1, M-CSF, CXCL10, IL-16, SERPIN E1/F1, CD105, OPN, and TF, with increased IGFBP5. Notably, CCL2, CXCL1, M-CSF, and GM-CSF—key MDSC recruitment factors—were downregulated.
RMC-6236 Induces Rapid Antitumor Responses in NRAS-Mutant Melanoma Mouse Models
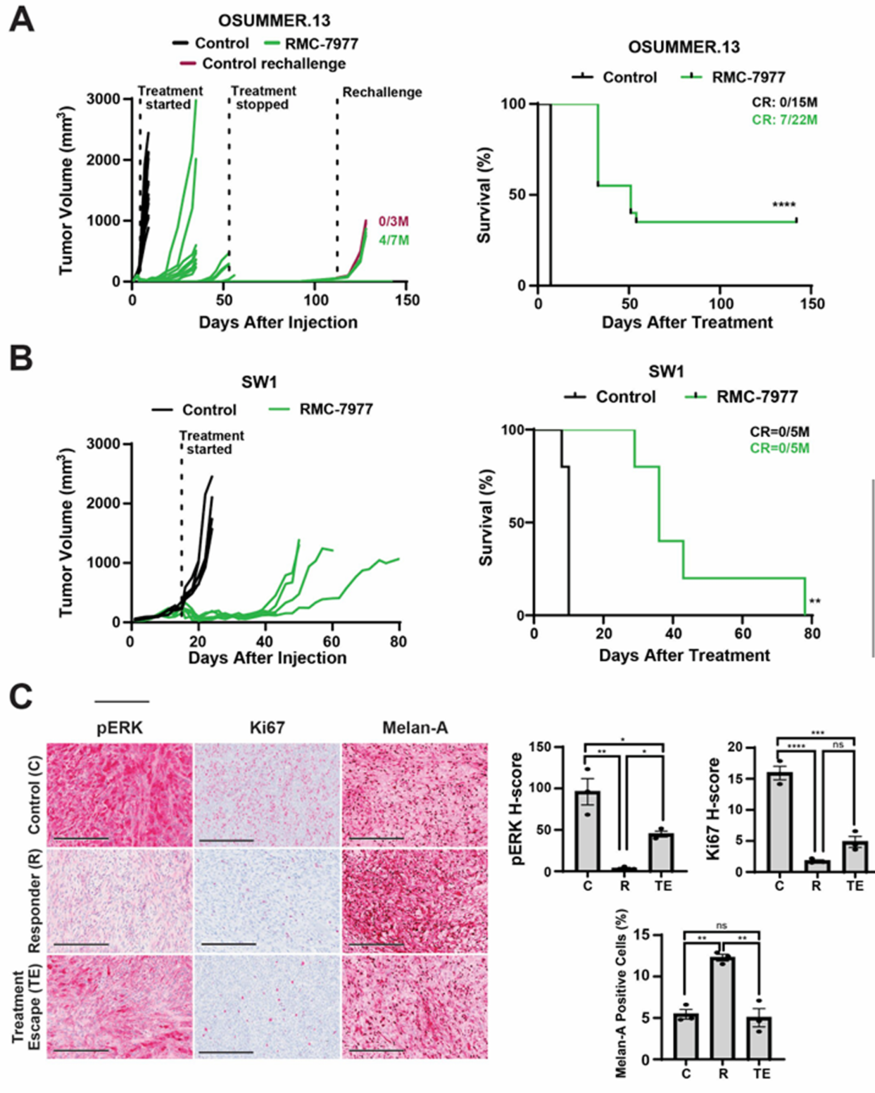
We tested RMC-6236 in mouse models, focusing on OSUMMER.13 (previously used for targeted/immunotherapy combination studies). Oral daily dosing (25 mg/kg) induced rapid tumor regression in all OSUMMER.13-bearing mice; some tumors regrew on therapy, while others remained tumor-free for 7 weeks. Complete regressions were durable post-treatment cessation. At day 112, rechallenging 7 tumor-free mice with OSUMMER.13 cells resulted in 4 remaining tumor-free, indicating immunological memory formation.
RMC-6236 was also potent in the SW1 model, inducing profound tumor regression and improving overall survival (though no complete regressions were observed). IHC analysis of vehicle-treated, early responder, and treatment-escape tumors showed: early OSUMMER.13 responders had reduced phospho-ERK and Ki67 (proliferation marker) and increased Melan-A (melanoma differentiation antigen); escape tumors showed recovered phospho-ERK/Ki67 and reduced Melan-A. In SW1 tumors, RMC-6236 decreased phospho-ERK/Ki67 and increased MHC-I/PD-L1.
RMC-6236 Upregulates MHC and Enhances T Cell Infiltration
Flow cytometry confirmed RMC-6236 upregulated MHC Class I, MHC Class II, and PD-L1 in responding OSUMMER.13 and SW1 tumors, with reduced expression in escape tumors (alongside MAPK pathway recovery). Immune microenvironment analysis revealed responding tumors had increased total/activated CD4+ and CD8+ T cells, enhanced CD4+/CD8+ memory T cells, and decreased Tregs. Escape tumors showed reduced activated T cells and increased Tregs—findings validated by IHC. Immune checkpoint expression (indicative of T cell activation) was higher in responders and lower in escape tumors. Collectively, RMC-6236 responses correlate with MAPK inhibition and enhanced T cell infiltration, while relapse links to MAPK recovery, reduced T cells, and increased Tregs.
RMC-6236 Alters Spatial Interactions Between MDSCs and T Cells
Multiplex immunofluorescence (9 markers: CD4+ T cells, CD8+ T cells, endothelial cells, M-MDSCs, PMN-MDSCs) confirmed flow cytometry/IHC results: responding tumors had increased CD4+/CD8+ T cells (waning with progression) and resistance correlated with M-MDSC accumulation. Spatial analysis showed responding tumors had dispersed MDSCs and T cells, while control/escape tumors had closer proximity between these cell types—suggesting suppressive cell-cell interactions in non-responding tumors.
Complete Responses to RMC-6236 Depend on CD4+ and CD8+ T Cells
Depletion studies clarified T cell roles: individual CD4+ or CD8+ T cell depletion did not affect initial RMC-6236 responses but abolished complete regressions. Combined CD4+/CD8+ depletion led to incomplete regression, early relapse, and shorter overall survival (no complete regressions). Mice without depletion had >50-day survival, with 7/15 achieving complete regressions. These data indicate RMC-6236's initial rapid antitumor effects stem from tumor pathway inhibition, but CD4+/CD8+ T cells are required to prevent resistance and achieve durable complete responses.
Anti-PD-1 + RMC-6236 Combination Boosts Durable Complete Responses
We tested whether anti-PD-1 (which reinvigorates exhausted T cells) enhances RMC-6236 efficacy. In OSUMMER.13 mice: anti-PD-1 monotherapy modestly delayed tumor growth; RMC-6236 monotherapy induced 2/7 complete responses; the combination was most effective (6/7 durable complete regressions) and significantly improved overall survival. Rechallenging combination-treated mice showed 5/6 remained tumor-free—indicating enhanced immunological memory vs. RMC-6236 alone.
IHC/flow cytometry showed the combination increased T cell infiltration, MHC-I/Melan-A expression, total/activated CD4+/CD8+ T cells, and memory T cells vs. monotherapies. It also reduced Tregs and exhausted CD8+ T cells (expressing PD-1, LAG-3, CTLA-4, or TIM-3)—a key improvement over RMC-6236 monotherapy (which increased Tregs and exhausted T cells). In the anti-PD-1-refractory SW1 model, the combination induced a minor but significant survival benefit. Efficacy gains correlated with the model's initial immunotherapy sensitivity.
RMC-6236 + Anti-PD-1 Reprograms the Tumor Microenvironment
Single-cell RNA sequencing (scRNA-Seq) of OSUMMER.13 tumors (vehicle, anti-PD-1, RMC-6236, combination) identified multiple immune cell types. RMC-6236 (alone or combined) drove larger transcriptional changes in the myeloid compartment than anti-PD-1, shifting tumor-associated myeloid cells from monocyte-dominant to macrophage/neutrophil-rich. Anti-PD-1 alone had minimal effects. Subclustering revealed increased antigen-presenting macrophages in anti-PD-1 and combination arms, with RMC-6236 inducing M1-polarized macrophages. The combination further increased M2 macrophages with antigen-presentation programs—indicating a mature antitumor immune response.
T cell compartment analysis showed the combination reduced naïve CD4+/CD8+ T cells, increased less-naïve/exhausted CD4+ T cells, and enhanced IFN-γ-expressing (cytotoxic) CD8+ T cells vs. monotherapies/vehicle. CellChat analysis (cell-cell communication) identified RMC-6236 as the major driver of microenvironment changes. The combination increased signaling strength between tumor cells and immune cells (neutrophils, NK cells, DCs, T cells, monocytes, macrophages, Tregs) and reduced tumor-macrophage communication vs. RMC-6236 alone. CXCL signaling was upregulated in RMC-6236-containing arms (stronger in combination), with key chemokines from tumor cells (CXCL1, CXCL12), neutrophils/macrophages (CXCL2, CXCL3), monocytes (CXCL2, CXCL9, CXCL16), and endothelial cells (CXCL2, CXCL9, CXCL12).
Daraxonrasib Demonstrates Clinical Activity in NRAS-Mutant Melanoma Patients
Two patients with NRAS-mutant melanoma were treated with daraxonrasib in a Phase I trial (primary endpoint: safety/tolerability; secondary endpoint: preliminary antitumor activity).
Patient #1: 77-year-old female with vulvar melanoma metastatic to lymph nodes, lung, and adrenal gland. She had progressive disease after pembrolizumab, ipilimumab+nivolumab, and pembrolizumab+axitinib. Tumor profiling showed NRAS Q61R, BAP1, FBXW7, and MAP3K4 mutations. She received daraxonrasib (300 mg/day, 21-day cycles) with doxycycline prophylaxis. Rapid improvement in a palpable inguinal mass was noted within 2 weeks; partial response (PR) was achieved after 2 cycles. Treatment was well-tolerated (mild toxicity). Restaging after 4 cycles showed ongoing PR in target lesions but progression in lung/adrenal/retroperitoneal metastases, leading to discontinuation.
Patient #2: 76-year-old female with heel melanoma, previously treated with excision, lymphoscintigraphy, adjuvant nivolumab, ipilimumab+nivolumab (brain metastases), stereotactic radiosurgery, nivolumab+relatlimab, lymphadenectomy, and radiotherapy. She developed in-transit/soft tissue metastases and nodal disease (RECIST v1.1 measurable). Tumor profiling showed NRAS Q61R (low tumor mutational burden). She received daraxonrasib (300 mg/day) but developed Grade 1 rash, nausea, fever, chills, and mucositis (managed with dose hold). After 2 cycles, target nodes achieved complete response (overall PR due to non-target lesions); complete response was confirmed after 4 cycles and maintained through cycle 6. Cycle 7 brought Grade 2 rash and mucositis (dose reduced to 200 mg/day, improving tolerance). Post-cycle 8 CT showed 66.7% increase in nodal target lesions (still <1.5 cm short-axis); treatment continued per protocol (ongoing clinical benefit, preserved performance status). Additional toxicities included Grade 2 diarrhea, rash, and palmar-plantar erythrodysesthesia. Disease progression was confirmed ~35 weeks post-initiation, and daraxonrasib was discontinued.
Discussion
RAS was long considered undruggable, but advances in KRAS inhibition (e.g., KRAS G12C inhibitors) have shown clinical promise. However, resistance to KRAS(OFF) inhibitors often arises from upstream RTK signaling that promotes RAS activation—highlighting the value of targeting active (GTP-bound) RAS to overcome such resistance.
Despite KRAS inhibitor progress, NRAS mutation-specific inhibitors remain limited. RMC-6236 and daraxonrasib (RAS(ON) multi-selective inhibitors) target active NRAS, HRAS, and KRAS. Preclinically, they show activity across KRAS-mutant cancers, with daraxonrasib demonstrating manageable safety and efficacy in previously treated pancreatic ductal carcinoma.
In this study, RMC-6236 potently inhibited RAS/MAPK signaling in NRAS-mutant melanoma (in vitro/in vivo), with partial phospho-ERK recovery unrelated to consistent RTK changes. It enhanced immune recognition (increased MHC-I/II, Melan-A, PD-L1) and reduced MDSC-recruiting cytokines (CXC3L1, IL-11, M-CSF, GM-CSF)—effects linked to MAPK inhibition (lost in escape tumors).
Oncogenic RAS drives immune suppression via cytokines (IL-1, IL-6, IL-8, CCL2, CXCL1) that recruit tolerogenic immune cells. RAS inhibition reverses this, invigorating antitumor immunity. For example, KRAS G12C inhibitor MRTX1257 increases IFN signaling and T cell recruitment in lung cancer. Consistent with this, RMC-6236 increased activated CD4+/CD8+ T cells and reduced M-MDSCs in NRAS-mutant melanoma models. Relapse correlated with MAPK recovery, reduced MHC expression, fewer T cells, and increased Tregs/MDSCs—with closer spatial proximity between T cells and MDSCs suggesting suppressive interactions (mirroring BRAF inhibitor resistance in melanoma).
While RMC-6236's initial antitumor effects were T cell-independent, immunological memory prevented tumor regrowth post-rechallenge. CD4+/CD8+ T cell depletion abolished complete responses, emphasizing adaptive immunity's role.
Combining RMC-6236 with anti-PD-1 potentiated responses: increased T cell infiltration, enhanced CD4+/CD8+ T cell differentiation, more IFN-γ+ cytotoxic CD8+ T cells, and reduced exhaustion. This was driven by enhanced melanoma antigen expression and myeloid cell antigen-presentation programs. scRNA-Seq showed RMC-6236 (alone or combined) altered myeloid cell proportions (increased macrophages/neutrophils) and induced M1-polarized macrophages—supporting a mature immune response. The combination also enhanced CXCL chemokine signaling, critical for immune cell recruitment.
In anti-PD-1-refractory models, the combination improved survival—paralleling findings in KRAS-mutant colon cancer. However, efficacy varies by model immunogenicity (e.g., "cold" tumors show limited response). Previous studies highlight the benefit of upfront immunotherapy followed by targeted therapy (improving MHC/antigen expression and immune priming)—a strategy supported by this study's combination data and clinical evidence in melanoma.
Preliminary clinical data showed daraxonrasib induced complete and partial responses in two heavily pretreated NRAS-mutant melanoma patients. The complete responder had a low tumor mutational burden and an isolated NRAS Q61 mutation, while the partial responder had a complex mutational profile. Biomarker studies are needed to identify predictors of response.
In conclusion, RAS(ON) multi-selective inhibition (e.g., daraxonrasib) shows preclinical and clinical activity in NRAS-mutant melanoma. Combining these agents with anti-PD-1 enhances antitumor immunity, offering a promising strategy for improving outcomes—particularly in ICI-resistant patients.
© 2026 ScienceTimes.com All rights reserved. Do not reproduce without permission. The window to the world of Science Times.










